
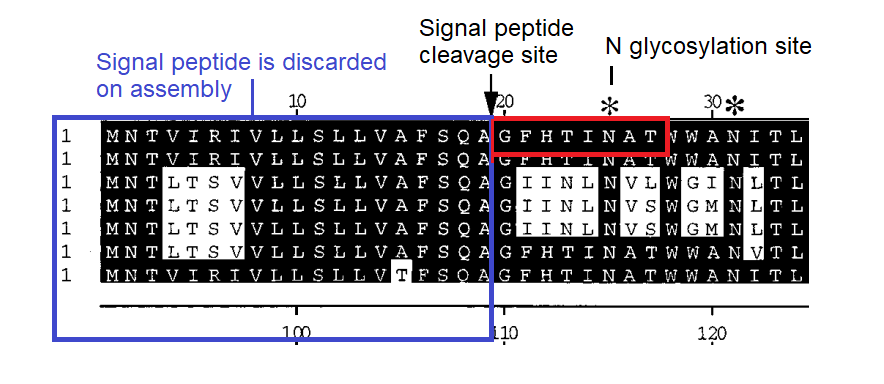
Early in the pandemic IIT scientists Pradhan et. al. attracted controversy when they identified 4 sites in the spike of SARS-CoV-2 as having inserts from HIV. I propose alternative explanations that are equally “uncanny”.
The Pradhan et al controversy marked the end of any semblance of reasonable discussion about pandemic origins and seems to have been the trigger for the infamous Fauci/Farrar teleconference. After this a lab origin became a “conspiracy theory” and a concerted effort was made to suppress discussion of the idea. Pradhan et al admitted their pre-print was written hastily, and perhaps didn’t get everything right. But they, quite sensibly, wanted to alert the world urgently to sequence anomalies they thought important for understanding pathogenesis and the possibility of an artificial origin.
They were criticized mostly because the regions of identity they found are small, and the corresponding regions in HIV are variable. HIV’s variable loops mutate even during the course of an infection within one individual (which is potentially decades long). So there are many possible HIV sequences these could match, and there wasn’t a single genome or region that was the source of their matches, but rather different regions in different genomes. Their hypothesis was perhaps over-reach, but it always came with caveats and disclosure.
But there are some things about which they were correct, that were lost in the shameful censorship and mockery that followed:
- These are all inserts compared to sequences known at the time. The only viruses having any similarity to those regions were the PLA published sequences ZC45 and ZXC21. However, since that time new sequences (starting with RaTG13) have been published that tend to obscure this fact, and, I suspect, intentionally.
- The regions do have homology with HIV-1’s variable loop regions, not only in the linear sequence, but structural and post-translational (e.g. N-glycosylation sites). This could also have suggested a natural explanation – convergent evolution. Sadly even this discussion was suppressed.
- Even if sequence identity to HIV-1 was coincidental it might have pointed to important viral functions (e.g. co-receptor binding, trafficking by immune system cells via DC-SIGN/L-SIGN, mechanisms of immune evasion via evolving patterns of glycosylation) and aided the development of safe vaccines and therapeutics.
Regardless of their origin, these sites have always been of interest to researchers. They have indeed proved to be evolutionary hotspots in the formation of new immune evasive variants, and there is evidence to suggest a possible role in co-receptor and/or sialic acid binding. Somewhat serendipitously I’ve found some surprising, but perhaps more plausible, explanations for their origin – in conserved regions of various distantly related human coronaviruses.
Assessing the probability of a sequence occuring “at random”
Prior to SARS-CoV-2 there were 6 known human coronaviruses (229E, OC43, NL63, HKU1, MERS, SARS). Each has a spike of ~1250 aas for a total of 8750 amino acids. This is small dataset, so finding a small sequence of common amino acids is within it is more significant than finding it within a variable HIV region (many thousands of sequences), let alone finding it in an unrestricted blast search of Genbank (which contains >10 trillion bases). It isn’t unexpected to find a simple 5 amino acid match between SARS-CoV-2 and another hCoV at random (we’d expect to find about one every 366 positions), in each case there are circumstances that make it far more unlikely, but calculating probabilities somewhat more difficult. Further complicating any discussion on probability is that evolution isn’t random, and also that all of these inserts occur at positions that are functionally important. So any numbers mentioned are just to give a rough idea.
NTD Loop 1: A mash-up of Adenovirus D and MERS?
I featured this region in the article on SARS-1, because it seems almost identical to the first few residues of the NTD of certain types (e.g. 37, 19) of an Adenovirus D protein found to be associated with sialic acid binding causing broad tissue tropism resulting in diverse clinical symptoms including conjunctivitis, corneal infection and pharyngitis.
Interestingly the significance of this protein was featured in a paper published just a few months before the first SARS case in 2002.
In SARS-1 the homology to Adenovirus 37 is striking, and improbable. It’s simple to calculate there are 25.6 billion possible combinations of 8 amino acids. A 7 out of 8 match is more frequent but still around 1 in 200 million. And that’s without the additional stipulation of a conserved N-glycosylation site in the final residues.

In SARS-CoV-2 some residues from the Adenovirus sequence remain, but there is also interesting homology to MERS, a region identified as forming part of its sialic acid binding pocket. The homology was first identified in early 2020 by a University of Warwick team):

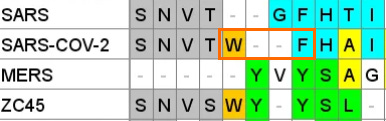
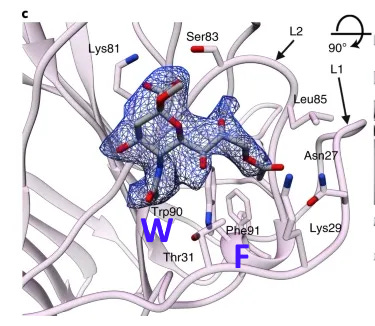
A paper from June 2019 paper (Tortorici et al). found a conserved sequence in coronaviruses that have been previously confirmed to bind sialic acids: OC43, BCoV, PHEV and HKU1. The sequence is different in MERS, but two aromatic amino acids – Tryptophan ![]() adjacent to a Phenylalanine (F) – occupy similar structural positions, despite being separate in the linear sequence. At the base of this loop in SARS-CoV-2 an adjacent W-F pair is formed, that is not present in MERS.
adjacent to a Phenylalanine (F) – occupy similar structural positions, despite being separate in the linear sequence. At the base of this loop in SARS-CoV-2 an adjacent W-F pair is formed, that is not present in MERS.
A curious pattern in ZC45
This Tryptophan ![]() is one of a couple shared uniquely with ZC45/ZXC21 at this site. These sequences are of interest for their provenance (being published by a largely PLA team from Nanjing Command in 2018). Notice also in ZC45 at this site the unusual number of doublets YY-SL-TT–NN–AA and the pattern of nucleotides, with the wobble base varying in each member of a pair.
is one of a couple shared uniquely with ZC45/ZXC21 at this site. These sequences are of interest for their provenance (being published by a largely PLA team from Nanjing Command in 2018). Notice also in ZC45 at this site the unusual number of doublets YY-SL-TT–NN–AA and the pattern of nucleotides, with the wobble base varying in each member of a pair.

Although the homology between ZC45 and SARS-CoV-2 isn’t exceptionally strong at this particular site, its significance is best understood in context of the other sites in the NTD, and some uniquely share mutations elsewhere in the genome (such as E and S2). Viewed in totality these suggest ZC45 maybe also an engineered virus (or fabricated sequence). This has important implications regarding the intentions of the engineers, suggesting a premeditated release of the virus, with ZC45/ZXC21 intended to provide evidence that such mutations had a precedent in nature.
NTD Loop 3: An ACE-2 binding region from NL63
*Numbering reflects position within the gene sequence, not the article.
This sequence is another extended exterior loop and also is an insert compared to related bat viruses. This sequence contains a 5 amino acid sequence PGDSS which is identical to one from unrelated human coronavirus NL63 – which as with SARS and SARS-CoV-2 uses ACE2 as its main receptor.


Once again there is also homology with ZC45/ZXC21, particularly in flanking residues, that is not seen in SARS or other bat SARS-like viruses known at the time.
But it isn’t just the 5 aas of sequence identity, 13 out of 14 residues in this region match, but in a different position, (including one conservative substitution (R→K). In a loop region which is intrinsically disordered, and contains many flexible residues (such as Serine (S) and Glycine (G)) the order in which they occur is likely less important.

Most important is the location of this sequence in NL63. According to a 2009 paper (Wu et. al.), which includes Fang Li as senior author, NL63’s RBM is composed of three loops separate in the sequence but adjacent in 3D structure. The SARS-CoV-2 NTD site matches the central loop of these (highlighted below). It’s also interesting that in SARS-CoV-2, three NTD loops that are separate in the sequence, come together in a similar way in the 3D structure.



This NTD loop also has homology with MERS (identified by Baker et al) in the residues at each end of the loop.

Though it is only 3 residues at this site it’s important to look at the 3D structural context, Sialic acid binds in a concave surface or “pocket”, the position of the active residues relative to the surface topology is crucial.
Looking first at Loop 1, the common residues between MERS and SAS-CoV-2 in this loop are coloured gold. The similarities here are obvious, in each case we have a G and T protruding like a finger, and an H, K and F in a recessed region at the base.

Though less easy to make out in a still image, the S, R and W residues from Loop 3 (coloured pink) also occupy structurally similar positions in the binding pocket (although in SARS-CoV-2 case the surface formed is concave but flat, not a hollow groove as in MERS).

NTD Loop 2: ZC45
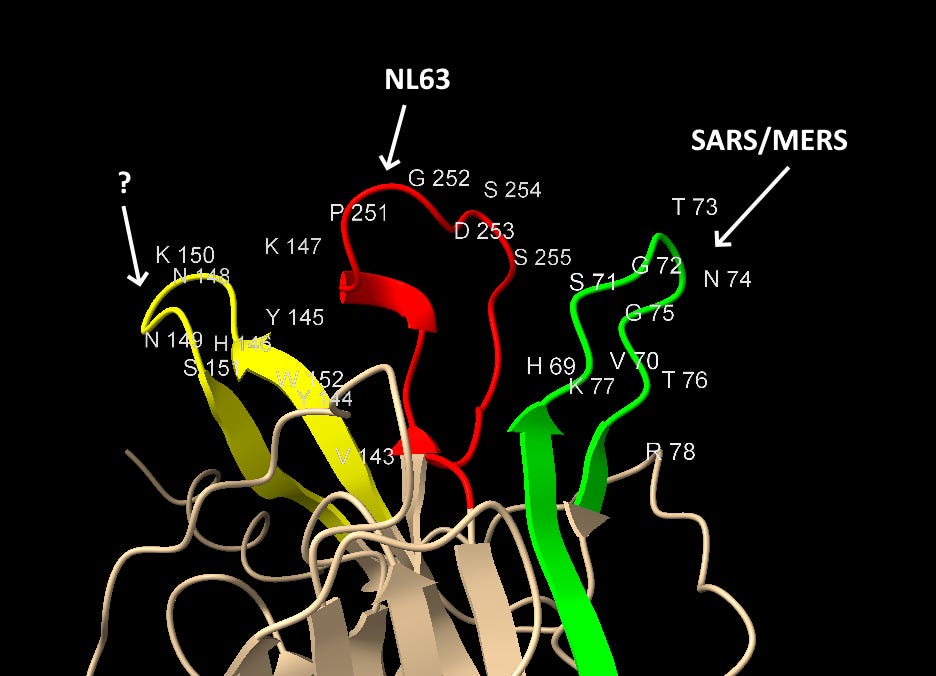
If our middle loop is derived from NL63, and the green loop is a SARS/MERS mash-up (via Adenovirus D), could the third loop also have homology to other human CoVs?
In this case the sequence aligns rather well with ZC45 – purportedly a bat, not human virus. The substitution S→T towards the 3’ is conservative and the N-glycosylation site is conserved

But, as I previously said, I have doubt that ZC45 is itself a natural virus, so could ZC45 have obtained this insert from elsewhere? The 4-mer sequence NNKS occurs in SARS-1 NTD at a different position. Just downstream is another NN pair which also creates a potential N-glycosylation site (or choice of):

Another curious coincidence I noticed in ZC45 is that all of the three corresponding NTD loops in its sequence contain an Asparagine pair NN and two contain a Tyrosine pair YY



I speculate that these sequences in ZC45 are experimental constructs based on observations made from the NTD of SARS-1. In this instance SARS-CoV-2’s corresponding region looks like a small refinement, whereas the others have been more radically reworked. But again there is some homology to MERS sequence in a structurally proximal loop, including the N-glycosylation site

Graphical Summary of NTD Loop Homology
The structures of SARS-CoV-2 and MERS are compared below with identical residues highlighted in red. SARS-CoV-2 contains many important residues from MERS’ sialic acid binding structure, in three different regions of the linear sequence, but coming together in the 3D structure.

Interspersed with the MERS residues, SARS-CoV-2 also has homology with other human coronaviruses SARS and NL63, and claimed bat coronaviruses ZC45/ZXC21. The NL63 homology is particularly unexpected, as the virus is unrelated, and the sequence comes from a different domain of the spike.
Discussion
While Pradhan et al may have found the wrong source for these inserts, they correctly identified suspicious locations in the SARS-CoV-2 spike. There are now many sequences that have been published since the start of the pandemic obfuscating the origin of these inserts, but also many reasons to ignore these as possible fabrications. I’ll substantiate this further in future articles.
I occasionally receive criticism that the kind of engineering needed sounds overly complex, but I’d point out that a grant application from Lanying Du states the following in the abstract:
“We use the RBDs from highly pathogenic coronaviruses, including MERS-CoV and SARS coronavirus (SARS-CoV), as the model system...we will construct chimeric RBDs containing the core subdomain from one coronavirus RBD as the structural scaffold and the RBM from another coronavirus RBD as the immunogenic sites”
Almost exactly as SARS-CoV-2’s NTD appears to be. More about this in the first article on the Furin Cleavage Site.
